Mathematics and the Family Tree of SARS-Cov-2
It's a remarkable process and, if it weren't so dangerous to humanity, would be deserving of admiration. ...
 Bill Casselman
Bill Casselman
University of British Columbia, Vancouver, Canada
Email Bill Casselman
Introduction
There are two major ways in which mathematics has contributed to our understanding of the disease CoVid-19 and the coronavirus SARS-Cov-2 that causes it. One that is very prominent is through mathematical modelling, which attempts to predict the development of the epidemic in various circumstances. In earlier Feature Columns I wrote about modelling measles epidemics, and much of what I wrote there remains valid in the new environment. With the appearance of the extremely dangerous CoVid-19, mathematical modelling has become even more important, but the complexity of this work has also become apparent, and maybe not suitable for discussion here.
Another relevant application of mathematics attempts to track the evolution of the virus responsible. The core of every virus is a small string of replicating genetic material, either DNA or RNA, which sits enclosed in a structure of some kind. The structure serves to keep the virus in one piece, but also, and most crucially, facilitates the insertion of the genetic material into target cells. (It is in this respect that SARS-Cov-2 outdoes SARS-Cov-1.) The genetic material then takes over the machinery of the invaded cell in order to reproduce itself. It's a remarkable process and, if it weren't so dangerous to humanity, would be deserving of admiration.
In larger animals, the basic genetic material amounts to double helices made up of two complementary strands of nucleotides---i.e., made up of the four nucleotides A (adenine), G (guanine), C (cytosine), and T (thymine). In the virus that produces CoVid-19 the genetic material is RNA, which in the larger cells plays little role in reproduction, but is involved in translating genetic instructions to the construction of proteins. It is a single strand of about 30,000 nucleotides, in which T is replaced by U (uracil). I'll ignore this distinction.
There is some variation in these strands among different populations of viruses, because strands that differ slightly can often still work well. The variation is caused by random mutations, which substitute one nucleotide for another, and a few other possibly damaging events such as deletions and insertions of genetic material. I do not know what, exactly, causes these, but some viruses are much more prone than others to mutate. For example, what makes the common influenza viruses so difficult to control is that they mutate rapidly, particularly in ways that make it more difficult to recognize them by antibodies from one year to the next. It's easy to believe that this "strategy" is itself the product of natural selection.
The good news is that coronaviruses do not apparently mutate rapidly. They do mutate enough, however, that RNA sequences of viruses from different parts of the world may be interpreted as a kind of geographical fingerprint. Keeping track of these different populations is important in understanding the spread of the disease. Similar techniques are also used to try to understand how SARS-Cov-2 originated in wild animal populations.
Where's the mathematics?
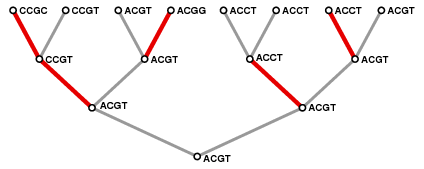
I'll try to give some idea of how things work by looking at a very small imaginary animal, with just four nucleotides in its genome. Every generation, it divides in two. In the course of time each of the progeny can suffer mutations at one site of the genome, and eventually each of these in turn divides. The process can be pictured in a structure that mathematicians call a
tree. In the following example, which displays the process through three generations, time is the vertical axis. The animal starts off with genome
ACGT. The colored edges mark mutations.
A structure like this is called a rooted tree by mathematicians and biologists. It is a special kind of graph.
And now I can formulate the basic problem of phylogenetics: Suppose we are given just the genetics of the current population. Can we deduce the likely history from only these data? The answer is that we cannot, but that there are tools that will at least approximate the history. They are all based on constructing a graph much like the one above in which genomes that are similar to each other are located close to one another. But how to measure similarity? How to translate a measure of similarity to a graph? You can already see one difficulty in the example above. The genome ACCT occurs in three of the final product, but although two of these are related in the sense that they are common descendants and therefore tied to the history, one is the consequence of independent mutations. It is difficult to see how this could be captured by any analysis of the final population.
It is this problem that the website NextStrain is devoted to. It is most distinguished for its wonderful graphical interpretation of the evolution of different populations of SARS-Cov-2. But, as far as I can see, it tells you next to nothing about underlying theory.
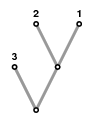
In the rest of this column, I'll try to give some rough idea of how things go. But before I continue, I want to come back to an earlier remark, and modify the basic problem a bit. I am no longer going to attempt to reconstruct the exact history. In reality, this is never the right question, anyway---for one thing, no process is a simple as the one described above. Reproduction is irregular, and one generation does not occupy steps of a fixed amount of time. Some organisms simply die without reproduction. Instead, we are going to interpret a tree as a graphical way to describe similarity, with the point being that the more similar two genomes are, the closer they are likely to be in our tree, and the more recent was their common ancestor. In particular, not all branches in our tree will be of the same length. The tree

illustrates only that A and B are more closely related than A and C or B and C. The actual lengths of edges in a tree will not be important, or even their orientation, as long as we know which node is the root. It is just its topological structure that matters.
Listing trees
All the phylogenetic methods I know of deal with trees that are
rooted and
binary. This means that (i) they all start off from a single node at the bottom and go up; (ii) whenever they branch, they do so in pairs. The endpoints of branches are called (naturally)
leaves. We shall also be concerned with
labeled trees in which a label of some kind is attached to every leaf. The labels we are really interested in are genomes, but if we number the genomes we might as well use integers as labels.
In order to understand some of the methods used to assemble relationship trees, we should know how to classify rooted binary trees (both unlabeled and labeled) with a given number of leaves.
There are two types of classification involved. First one lists all rooted binary trees of a given shape, and then one looks at how to label them. For a small number of leaves both these steps are very simple, if slightly tedious.
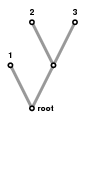
Two leaves. They all look like the figure on the left:
And there is essentially one way to label them, shown on the right. What do I mean, "essentially"? I'll call two trees essentially the same if there exists a transformation of the one of them into the other that takes edges to edges and the root to the root. I'll call two labelings the same if such a transformation changes one into the other. In the tree above the transformation just swaps branches.
Three leaves. There is only essentially one unlabeled tree. To start with, we plant a root. Going up from this are two branches. One of them must be a leaf, and the other must branch off into two leaves. So the tree looks essentially like this:
As for labelings, there are three. One must first choose a label for the isolated leaf, then the others are `essentially' determined:
(There are three other possible labelings, but each yields a tree essentially the same as one of those above.)
Four leaves. There are two types of unlabeled trees, one with branching to an isolated leaf and an unlabeled tree of three leaves, the other with two branches of two leaves each.
There are 15 labelings. There is a systematic way to list all rooted trees with $n$ leaves, given a list of all those with $n-1$, but I won't say anything about it, other than to hint at it in the following diagram:
I will say, however, that there are 105 labeled trees with 5 leaves, and that in general, there are $1\cdot 3 \cdot \, \ldots \, \cdot (2n-3)$ labeled trees with $n$ leaves. (I refer for this to Chapter 2 of the lecture notes by Allman and Rhodes.) This number grows extremely rapidly. It is feasible to make lists for small values of $n$, but not large ones. This impossibility plays a role in reconstructing the phylogeny of a large set of viruses.
Reconstruction
Reconstructing the entire history in practical cases is for all practical purposes impossible. What one attempts is just to find an approximation to it. The structure of the candidate graph should reflect at least how close the items are to each other. As explained in the lecture notes by Allman and Rhodes, there are many different ways to measure closeness, and many different techniques of reconstruction. None is perfect, and indeed there is no perfect method possible.
The techniques come in two basic flavours. Some construct a candidate tree directly from the data, while others search through a set of trees looking for the one that is best, according to various criteria. The ones that construct the tree directly don't seem to do well, and the methods most often used examine a lot of trees to find good ones. I'll look at the one that Allman and Rhodes call parsimony. It is not actually very good, but it is easy to explain, and gives some idea of what happens in general. It tries to find the tree that is optimal in a very simple sense---it minimizes, roughly, the total number of mutations in terms of the connections of the tree. (The term, like many in modern biology, has taken on a technical meaning close to, but not quite the same as, that in common usage. According to Wikipedia, parsimony refers to the quality of economy or frugality in the use of resources.)
In this method, one scans through a number of rooted trees whose labels are the given genomes, and for each one determines a quantitative measure (which I'll call its defect) of how closely the genomes relate to each other. One then picks out that tree with the minimum defect as the one we want. In fact, in this method as in all of this type there may be several, more or less equivalent.
How is the defect of a candidate labeled tree computed? I'll illustrate how this goes for only one set of genomes and one tree:
Step 1. The defect of the tree is the sum of defects of each of its nucleotides (in our case, four). This is done by calculating at every node of the tree a certain subset of nucleotides and a certain defect, according to this rule: (i) if the node is a leaf, assign it just the nucleotide in question, and defect 0. Progress from leaves down. At an internal node whose branches have already been dealt with, assign a subset --- either (i) the intersection of the subsets just above it if it is non-empty, or (ii) otherwise, their union. In the first case, assign as defect the sum of defects just above, but in the second assign the sum plus 1. These four diagrams show what happens for our example.
The total defect for this labelled tree is therefore 4.
Step 2. In principle, you now want to scan through all the trees with $n$ leaves, and for each one, calculate its defect. There will be one or more trees with minimal defect, and these will be the candidates for the phylogeny. In practice, there will be too many trees to look at all, so you examine a small collection of sample trees and then look at a few of their neighbours to see if you can make the defect go down. The trick here, as with many similar methods is to choose samples well. I refer to Chapter 9 of Allman-Rhodes
Dealing with SARS-Cov-2
You can test a method for reconstructing an evolution by running some evolutionary processes and then applying the method to see how it compares. In practice, the structure parsimony produces are not generally satisfactory. In practice, it is the method "Maximum Likelihood" used most often. Allman-Rhodes say something about that method, too. As with every one of the methods so far developed, there are both theoretical and practical difficulties with it. But in most situations its results seem to be satisfactory.
With SARS-Cov-2, there are at least very satisfactory sources of data --- GenBank and GISAID. I say something about these in the reference list to follow. Both of these websites offer more than thousands of genomes for SARS-Cov-2 as well as other organisms. These have been contributed by medical researchers from all over the world, and they are well documented.
Mathematics
Biology
- The second edition of James Watson's Molecular Biology of the Gene.
Chapter 15 is a lucid (and terrifying) account of what was known at the time it was written about what a virus does inside a victim. (I have not had access to more recent editions.)
- The first published genetic analysis of SARS-Cov-2
- Parsimony
- Nextstrain is a deservedly famous project that tracks, among other things, the evolution of strains of SARS-Cov-2 with impressive graphics. It allows the user to examine developments interactively, and includes also a collection of tools to carry out your own analysis (i.e. DIY genetics). However, as far as I can see it is weak in explaining the theory behind their tools.
- GenBank
Open policy, very convenient, but less complete than GISAID (below).
- GISAID
This is the most complete archive of SARS-Cov-2 genomes, but with somewhat restrictive rules about publishing the data found there. It requires registration for access. This is not surprising---there are potentially an enormous number of fortunes to be made.
- University of Washington software list
To a mathematician, the sheer volume of publications on medical topics is astonishing. I list this site to illustrate this even for the relatively arcane topic of phylogeny. Incidentally, the original reason for developing phylogenetic tools was to track the evolutionary development of life on Earth, which was a matter of interest even before Darwin's theory of evolution appeared. For many years, it was based on morphological aspects rather than genomes. But now that gene sequencing is so simple, it has also become an important part of medical investigation.
Bill Casselman
University of British Columbia, Vancouver, Canada
Email Bill Casselman